





Author: Yervant Chijian, Director, Team Lead Medical Devices / IVD Australia, PharmaLex
As with any other medical device product, manufacturers of medical software products are required to manage the life cycle of their medical device software according to defined processes and regulatory requirements.
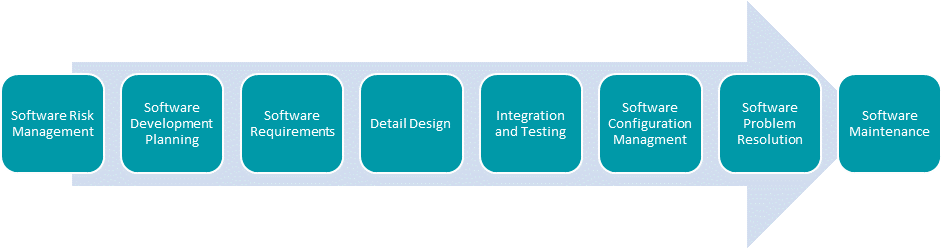
To achieve these requirements, companies need to follow the relevant standards, and specifically the IEC 62304, which specifies the life cycle processes, activities, tasks, and deliverables required to develop medical device software including both Software in a Medical Device (SiMD) and Software as a Medical Device (SaMD).
International standards have been designed for the purpose of assisting manufacturers with the processes required for managing and maintaining the integrity of their products and leverage industry best practices. Therefore, IEC 62304 establishes a best practices framework for software development, while a separate standard, ISO 14971 defines the processes to reduce and manage risks with medical devices.
 Key Elements to Medical Device Software Life Cycle Management
Key Elements to Medical Device Software Life Cycle Management
Integral to the life cycle management of a SaMD product is establishing a quality management system (QMS) design. Internationally, there are standards and regulations that provide a framework for implementing a QMS based on best practices, such as ISO 13485 and the IMDRF SaMD application of the QMS draft guidance. However, certain subsystems within a QMS need to be interpreted according to the software lifecycle processes, as software itself has a unique development process, with a key differentiator being the fact that there is no physical product, and therefore many QMS manufacturing processes do not apply for software. Specific software development procedures should be established based on the IEC 62304 standard and integrated within the ISO 13485 processes.
An effective software development plan should be developed within a QMS framework early in development. Verification and validation planning need to be considered within the project, should be scalable for the type of software and the size of the organization and should take into consideration important elements to show safety, effectiveness and performance. Procurement and supply controls in both outsourced processes and use of commercial off-the-shelf software is needed to deliver safe and effective software. This is crucial as software developers increasingly rely on either outsourcing the software development or integrating off-the-shelf software products into their code. Regulators require the manufacturer to have a level of control of any third-party software integrated within their device.
Design Verification and Validation Process
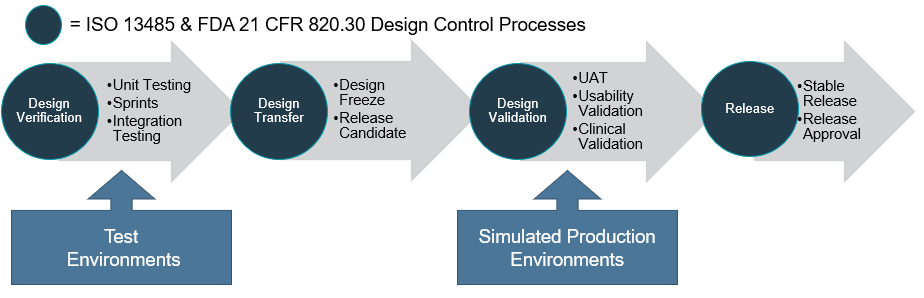
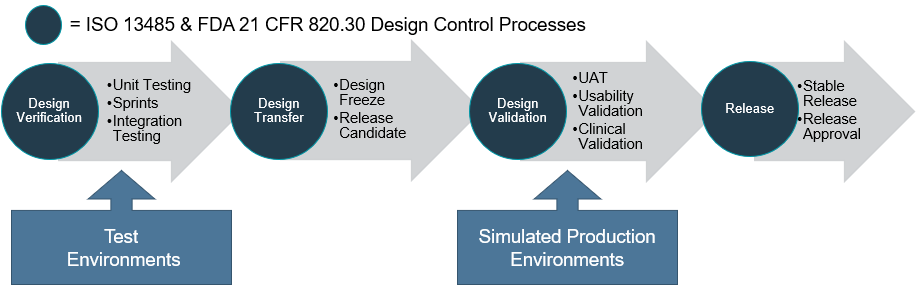
Design verification involves unit testing, integration testing and can be conducted in an iterative process. Once all the software requirements have been implemented and verified, a design freeze is approved allowing a release candidate to be ready for design validation during the design transfer phase.
The design validation may then involve user acceptance testing, usability validation and clinical validation. At the completion of the design verification and validation process, the final software is approved for release as a stable build to the corresponding production platform or commercial use environment and any changes should be managed through software version and change control procedures.
 Verification and Validation Design Process in the Context of Software Development
Verification and Validation Design Process in the Context of Software Development
Typically, during software development, various environments are used for testing. An important point here is to ensure that the environment for design validation is the same as the production platform or an equivalent simulated platform is used.
Usability engineering in development
SaMD manufacturers also should adopt usability engineering processes, based on international standards (such as IEC 62366-1), early in development to ensure their product achieves reasonable usability and to minimize user errors and associated user risks.
Usability engineering begins during the design phase with defining the preliminary user interface and user experience (UI/UX) requirements. This allows manufacturers to identify potential hazards, such as user errors and foreseen misuse.
During the detailed design and output phase, the UI/UX requirements are converted to engineering specifications, such as the software requirements specifications, and implemented within the software code or interface design. When designing the preliminary UI/UX, there needs to be consideration about the technical knowledge, experience, education and training, and, where applicable, the medical and physical abilities of the intended users.
During the verification and validation (V&V) phase, the UI/UX design is verified along with the software V&V processes. User evaluations, formative or summative studies involving study participants, and findings from these studies typically are contained in a usability study or evaluation report.
Another key consideration with software verification and validation is the use of operating platforms. Since SaMDs can be deployed on various operating platforms, such as desktop PC apps, mobile apps, web apps and software as service, the manufacturer must ensure the platform specifications are defined during product development and that the SaMD is validated on the specified target platform or equivalent before release.
Post-marketing surveillance considerations
Once the software is produced and released, regulators expect manufacturers to collect and analyze post-marketing data for the life of the device. Due to the flexible nature of software, it is possible to incorporate mechanisms within the software to collect that data for regular monitoring and review.
There are four key post-marketing activities that SaMD manufacturers must follow. These include:
- Issue management, covering processes for adverse event monitoring, complaints and record processing, problem resolution processing.
- Information security or cyber security, covering processes for monitoring vulnerabilities and threats and reducing the risk of attacks.
- Software maintenance, applying standard software processes for managing user requirement changes and enhancements, managing third party software changes, managing configuration changes, for example platform or OS changes.
- Quality assurance, either built into the software or defining processes to gather customer feedback. Specifically, building into the design functions the ability to gather quality metrics such as quality of input or process data.
During each of these processes, the risk and clinical regulatory impact should be assessed for any impact on the regulatory status of the software to ensure that the software will continue to operate as intended and that there are no hazards that could directly or indirectly harm the patient or change the classification status.
Artificial Intelligence and Machine Learning
The software development process is becoming even more critical with artificial intelligence and machine learning (AI/ML) algorithm changes, which could vary the use of the SaMD, the intended use and regulatory status. This has posed challenges for regulators, and in 2021 the US Food and Drug Administration issued its Artificial Intelligence/Machine Learning (AI/ML)-Based Software as a Medical Device (SaMD) Action Plan.
The plan would seek a commitment from manufacturers on transparency and real-world performance monitoring and periodic updates on changes implemented. The objective is to enable oversight without undue restriction of iterative improvements enabled by AI or machine learning-based software, while safeguarding the patient. This would require manufacturers to develop algorithm change control plans defining the boundaries for the AI/ML for changing the algorithm after validation and release, and criteria for revalidation with real-world data.
Embracing standards and differences
While SaMDs differ in nature from other medical devices, international standards offer strong guidelines for managing the design, life cycle management, risk management and surveillance of SaMDs.
Adopting the IEC 62304 standard for the software development lifecycle provides manufacturers with the essential framework required by most regulators to develop medical device software. The standard defines the processes that can be integrated into the manufacturer’s QMS to ensure that the required deliverables are generated during development. Therefore, it is critical that the software development procedure be developed prior to the development of the software. Failure to do so would require retrospective creation of documentation and other activities such as risk assessments or usability evaluations, which may lead to unexpected design changes, delays in release of the software and commercial delays to market access.



