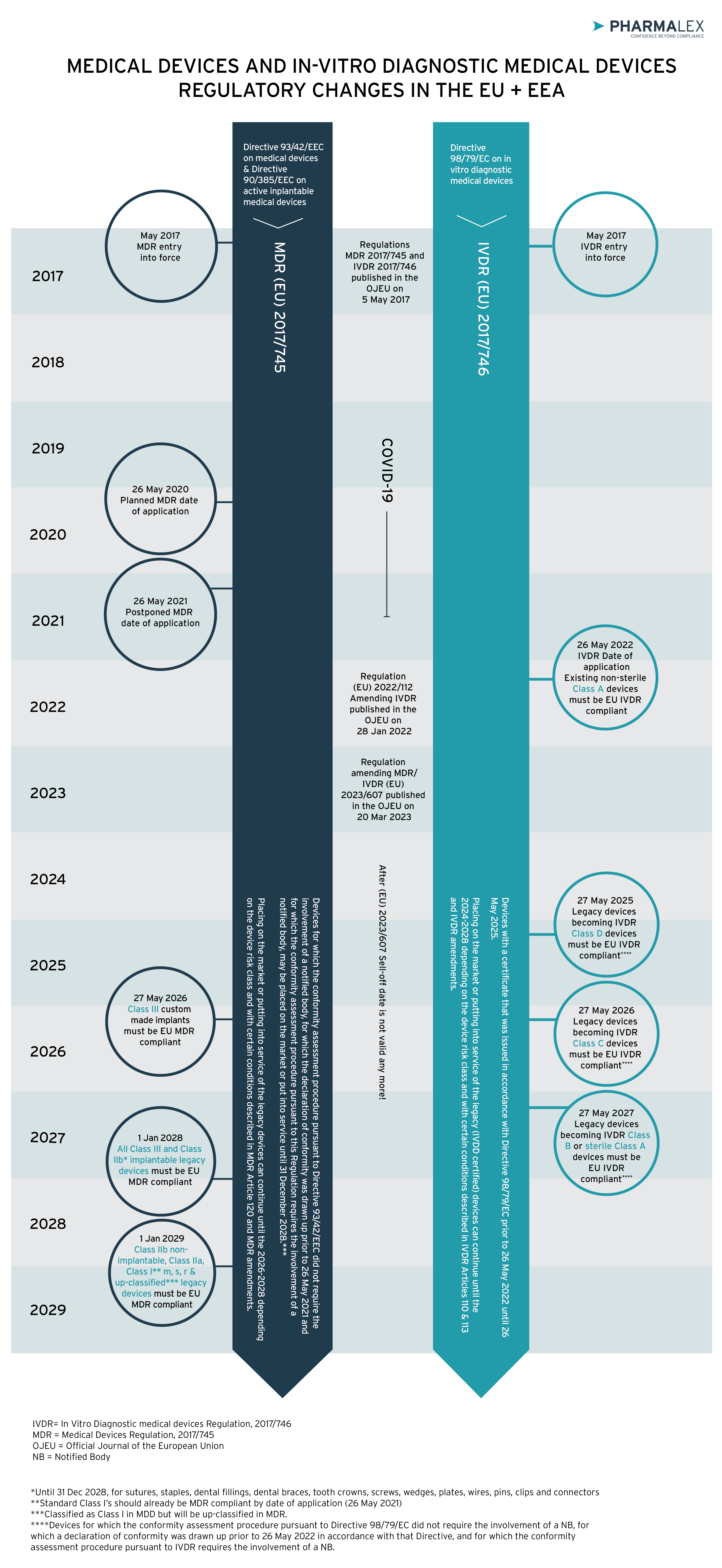
Since 2017, there have been numerous regulatory changes in the European Union (EU) related to medical devices and in vitro diagnostics, including the Medical Device Regulation [MDR (EU) 2017/745] (“MDR”) and In Vitro Diagnostic Medical Devices Regulation [IVDR (EU) 2017/746] (“IVDR”). More recently, the EU introduced regulation which mostly amended MDR but also partially affected IVDR. Regulation (EU) 2023/607 was published in the Official Journal of the European Union on 20 March 2023 and it became applicable with immediate effect.
There is a lot of confusion about what is required given the many recent regulatory changes. Having clear insight into what these changes are and what they mean will help economic operators — manufacturers, importers and distributors — to ensure they are ready to meet the regulations.
PharmaLex has collected two checklists with main points highlighting the practical meaning of the latest changes and what these mean to key stakeholders. The Manufacturers’ Checklist includes issues relevant to manufacturers. Similarly, the Importers and Distributors Checklist includes topics that are relevant for those economic operators.
In the accompanying flow chart you can see an overview of what has happened in Europe’s medical devices and in vitro diagnostic medical devices regulatory area during the previous years.
References:
Regulation (EU) 2023/607 of the European Parliament and of the Council of 15 March 2023 amending Regulations (EU) 2017/745 and (EU) 2017/746 as regards the transitional provisions for certain medical devices and in vitro diagnostic medical devices.
Consolidated text: Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and repealing Council Directives 90/385/EEC and 93/42/EEC. (20/03/2023)
Consolidated text: Regulation (EU) 2017/746 of the European Parliament and of the Council of 5 April 2017 on in vitro diagnostic medical devices and repealing Directive 98/79/EC and Commission Decision 2010/227/EU. (20/03/2023)
Regulation (EU) 2022/112 of the European Parliament and of the Council of 25 January 2022 amending Regulation (EU) 2017/746 as regards transitional provisions for certain in vitro diagnostic medical devices and the deferred application of conditions for in-house devices.
MDCG 2021-25 Regulation (EU) 2017/745 – application of MDR requirements to ‘legacy devices’ and to devices placed on the market prior to 26 May 2021 in accordance with Directives 90/385/EEC or 93/42/EEC.
MDCG 2022-8 Regulation (EU) 2017/746 – application of IVDR requirements to ‘legacy devices’ and to devices placed on the market prior to 26 May 2022 in accordance with Directive 98/79/EC.
Template for notified body confirmation letter of the status of a formal application, written agreement, and appropriate surveillance in the framework of Regulation EU 2023/607.
MDCG 2022-18 MDCG Position Paper on the application of Article 97 MDR to legacy devices for which the MDD or AIMDD certificate expires before the issuance of a MDR certificate.
EU Commission QA document “Extension of the MDR Transitional period and removal of the ‘sell off’ periods” – Q&A on practical aspects related to the implementation of Regulation (EU) 2023/607 amending Regulations (EU) 2017/745 and (EU) 2017/746 as regards the transitional provisions for certain medical devices and in vitro diagnostic medical devices, March 2023.
Consolidated text: Directive 98/79/EC of the European Parliament and of the Council of 27 October 1998 on in vitro diagnostic medical devices
Consolidated text: Council Directive 93/42/EEC of 14 June 1993 concerning medical devices
Consolidated text: Council Directive of 20 June 1990 on the approximation of the laws of the Member States relating to active implantable medical devices (90/385/EEC)
Flowchart to assist in deciding whether or not a device is covered by the extended MDR transitional period