






Author: Rob Stringer
The standard regulatory evaluation pathway in Australia for new prescription medicines involves a thorough TGA review of data demonstrating safety, efficacy +/- quality. In addition to the standard regulatory pathway for new or extended uses of prescription medicines in Australia, the TGA also offers several different regulatory pathways to market for applicants to consider.
Provisional Registration Pathway
The provisional registration process allows certain medicines to be provisionally registered in the Australian Register of Therapeutic Goods (ARTG) for a limited duration. These medicines are registered on the basis of preliminary clinical data, where the benefit of early availability of the medicine outweighs the risk inherent in the fact that additional data are still required.
- Products must be accepted into the pathway prior to submission of a registration application, and all approved provisional designations are published on the TGA website here.
- The provisional pathway allows for the submission of early data with a standard review timeline (~15 months). Provisional approval is conditional upon submitting additional data to transition to full regulatory approval.
- Application and evaluation fees are higher for the provisional registration process than for the standard prescription medicines registration process. The regulatory process can allow for a limited number of rolling submissions of clinical data during the evaluation phases.
- The regulatory review involves closer coordination between clinical, nonclinical, quality and Risk Management Plan (RMP) evaluation areas to ensure that any emerging safety signals are identified and managed (to assist with managing the risks of preliminary clinical data). Additionally, the TGA may request an expert Advisory Committee throughout the regulatory process.
- Indicators for eligibility for provisional review can be gleaned from other, similar designations granted by other agencies, e.g. EMA conditional review, and US FDA accelerated approval.
More information on the provisional process is available here.
Priority Review Pathway
The priority pathway provides a formal mechanism for faster assessment of vital and life-saving prescription medicines. The priority pathway is suitable for products with a strong unmet need where there are no other treatments available and/or the product is better than existing registered products.
- In Australia, applicants must seek approval of a priority determination for their application prior to submission of a registration application.
- The eligibility criteria for priority determination are designed to ensure that only medicines providing the most benefit to patients are approved. All approved priority determinations are published on the TGA website here.
- Priority review is based on a full dossier and substantial evidence. The target timeframe of 150 working days is up to three months shorter than the standard prescription medicines registration process.
More information on the priority determination process and pathway is available here.
Comparable Overseas Regulator (COR) Pathways
The COR pathway came into effect in January 2018 as a TGA initiative to reduce evaluation review timelines based on leveraging another regulator’s approval and evaluation documentation.
The following countries can be used as the COR regulator for such applications, with the onus upon the applicant to directly provide the TGA with unredacted evaluation documentation from the corresponding review.
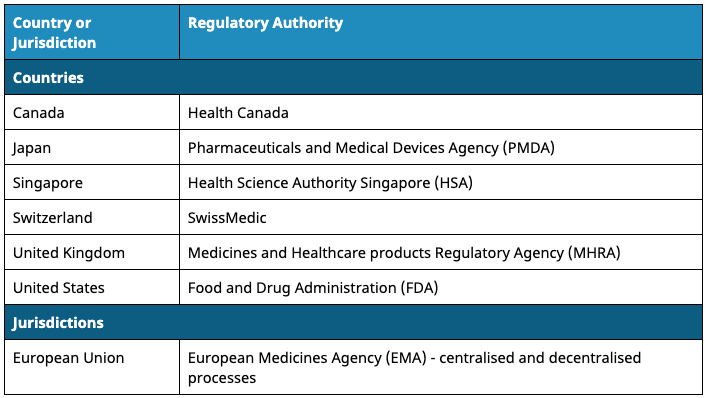
There are two options within the COR pathway regulatory process:
- COR A Pathway – 120 working day evaluation from acceptance into evaluation
Submission must be within 12-months of the COR regulator’s approval. Applicants need to apply for a consistent indication to that which was approved by the COR agency and, all the manufacturing detail information must be identical to that approved for the COR review. - COR B Pathway – 175 working day evaluation from acceptance into evaluation
No time limit within which the application can be submitted; certain manufacturing changes are permitted.
Australia-Canada-Singapore-Switzerland-Unit Kingdom (Access) Consortium
The Access Consortium was formed in 2007 to promote greater regulatory collaboration and alignment of regulatory requirements. The Access Work Sharing Initiative has resulted in the approval of several medicines through this international collaboration reducing regulatory duplication and increasing each agency’s capacity to ensure timely access to medicines.
The Access Consortium pathway can be utilised for New Chemical/Biological entities:
- Requires alignment of submission timelines and pathways for participating countries to enable regulators to work-share the evaluation of data. Region-specific commercial agreements may limit/complicate ability to participate.
- Timeline is typically quicker than the standard regulatory process with applicants receiving consolidated sets of questions (thereby reducing the burden on global teams)
Applications that have successfully received market approval through this initiative include: Baloxavir marboxil, Niraparib, Abemaciclib and Apalutamide. 1
Project Orbis
Project Orbis is an initiative of the FDA Oncology Centre of Excellence (OCE). This initiative provides a framework for concurrent submission and review of oncology products among international regulators. 2 Project Orbis has facilitated collaboration amongst selected other agencies including Australia, Canada, Switzerland, Singapore, Brazil and the UK.
US Companies can be invited by the US FDA OCE to participate in Project Orbis and/or propose applications and the OCE acts as the primary coordinator for the process. Acceptance into the initiative is contingent upon meeting key clinical criteria (for example clinically significant and/or compelling data demonstrating improved safety or efficacy) and a commitment by the applicant to be able to facilitate the submission of applications in close proximity in all participating territories. Once a viable applicant/pathway for regulatory filing has been identified for each of the intended jurisdictions, the FDA will liaise directly with the respective regulatory agencies to inform them of the potential for evaluation under the Project Orbis framework. Once participation by the agency has been confirmed, the application can proceed to the next stage. More information on Project Orbis is available here.
There are a number of considerations to take into account when selecting the optimal pathway to market. Please contact the team for assistance today.


