Since Regulation (EU) 2017/745, also known as the MDR, came into force, a number of products without an intended medical purpose but that have similarities with the functionality of certain medical devices have been impacted. A concept the European Commission refers to as common specifications now covers these products via a draft Commission Implementing Regulation released in December 2022.
The EU MDR defines common specifications as “a set of technical and/or clinical requirements, other than a standard, that provides a means of complying with the legal obligations applicable to a device, process or system”.
Products affected by these common specifications are described in Annex XVI and include:
- Contact lenses with aesthetical purpose (e.g., on-prescription coloured contact lenses).
- Products intended to be introduced into the human body through invasive surgery (e.g., surgically implanted permanent lip implants).
- Substances intended to be used for facial or other dermal or mucous membrane (e.g., dermal fillers)
- Equipment intended to be used on adipose tissue (e.g., liposuction equipment)
- High intensity electromagnetic radiation emitting equipment intended for use on the human body (e.g., Intense pulsed light machines for body hair removal)
- Equipment intended for brain stimulation with magnetic or electromagnetic fields (e.g., transcranial (non-surgically invasive) stimulation).
There are important steps to consider in order to comply with the legal requirements.
First, the product should be classified according to Annex VIII of the MDR.
A technical file should be prepared to provide evidence of compliance with the common specification and MDR requirements.
For these types of products, the manufacturer will need to demonstrate compliance with the common specifications. These common specifications are technical and clinical requirements as well as risk management measures that allow compliance with the safety and performance obligations of these devices.
For products classified as class I sterile or with measuring function, IIa, IIb and III, a notified body should be selected to conduct the CE certification process.
Where compliance with the applicable requirements has been demonstrated, a CE declaration of conformity should be issued in accordance with Annex IV.
A Basic Unique Device Identifier (Basic-UDI) should be assigned and information on economic operators and products should be submitted in Eudamed.
Once all these steps have successfully been completed, the product can be placed on the EU market.
Obligations continue even after the product is on the market, and, as with all medical devices, the manufacturer should meet the post-market surveillance and vigilance requirements throughout the product lifecycle.
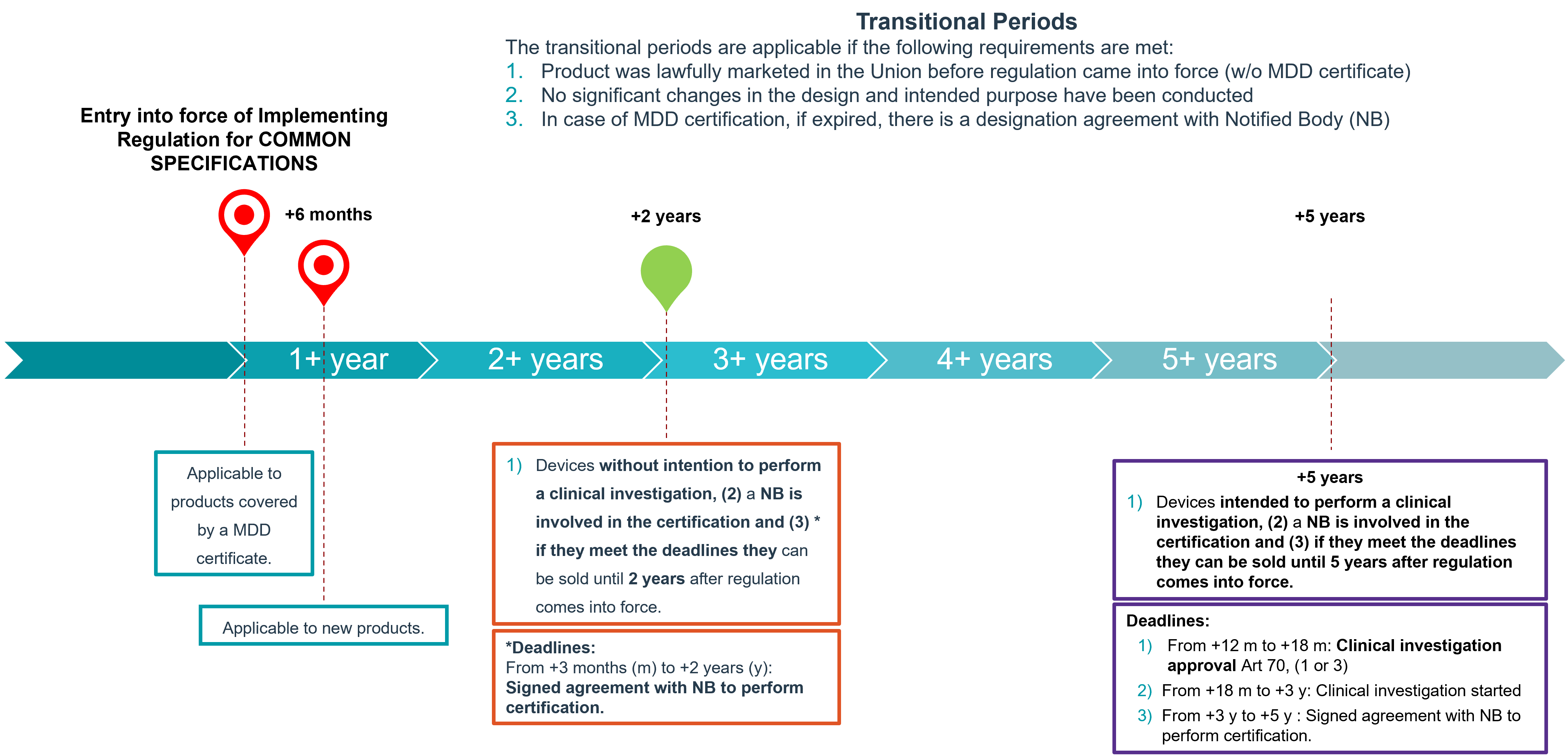
When will these common specifications be applicable?
The final document defining the common specifications for Annex XVI products is still pending, however information can be found here. These common specifications will be applicable 6 months after the Commission Implementing Regulation comes into force, except for the products covered by a certificate in accordance with Directive 93/42/EEC.
The application of the implementing regulation and the transitional provisions are detailed in the following diagram: