





Defining and Regulating the Complex World of Software as a Medical Device
The medical devices landscape has changed dramatically in recent years with the shift from hardware-based medical devices to the advent of smart, standalone software technology medical devices. However, the definition of what constitutes software as a medical device (SaMD) is not that simple and that poses some unique challenges when it comes to regulations. Furthermore, since regulations were written well before the emergence of SaMDs, health authorities have had to consider how to adapt these regulations to the fast-moving nature of the technologies.
The use of software in medical devices has grown exponentially and in different ways. However, SaMDs are a distinct class, separate from what is referred to as software in a medical device (hardware incorporating software as an integral part of its function) and software used to help manufacture or maintain a medical device. The introduction of the SaMD class brings with it a new group of innovators and manufacturers, many of whom have no or very little experience with regulations and are struggling to get up to speed with the different global regulatory requirements and what they mean for their products.
The regulatory landscape
Let’s explore the key regulatory challenges that are triggered by these evolving technologies and the steps regulators and standards bodies are taking to address these challenges.
A product is defined as an SaMD if the software is standalone and meets the definition of a medical device, which is that it is intended to be used for diagnosis, screening, prevention, monitoring, treatment or alleviation of disease.
While the nature of SaMDs is complex and there are inevitably different interpretations in different jurisdictions, there are efforts to harmonize regulatory approaches. The International Medical Device Regulators Forum (IMDRF), a voluntary group of medical device regulators, formed the Software as a Medical Device Working Group aimed at encouraging innovation and access to SaMDs by establishing key definitions, a risk categorization framework, quality management system principles, and a clinical evaluation pathway.
A SaMD will be regulated in the same way as other medical devices of the same risk category, however, the classification rules that exist for medical devices currently only consider the possible harm caused by physical interaction of the medical device for human. As software does not have this physical interaction, the risk associated with these types of products relate more to the information provided by the SaMD fir diagnosis and clinical treatment recommendations, such as analysis and calculation errors, inaccuracies, and poor user interfaces (UI/UX). The risk classification from these potential inaccuracies have been laid out by the IMDRF depending on the type of condition it supports. These include:
- Non-serious, where interventions are normally non-invasive, giving the user an opportunity to detect flawed recommendations. Users can either be specialized users or lay users
- Serious, where disease progression is moderate but where major therapeutic intervention isn’t required or time critical. Again, users can either be specialized users or lay users
- Critical, where the health of the patient is life-critical and where intervention can be time critical, meaning the user may not be in a position to reflect on recommendations. In these circumstances, only specialized, trained users should interpret the SaMD information.
Exploring the regulatory frameworks
The next question manufacturers might have is, how do these categories affect the way a product is regulated?
At a high level, there are two main regulatory frameworks for SaMDs, as there are with hardware devices. The first is the GHTF framework, which encompasses most jurisdictions beyond the United States – Europe, Australia, Canada, Japan and others. The GHTF framework relies on the compliance with a set of essential requirements for safety and effectiveness, as opposed to clinical experience for the demonstration of safety and effectiveness.
The second framework, adopted by the U.S., adopts a premarket submission approach where classification is based on product codes, which are contained in FDA’s Product Code catalogue for the various device types. The class classification found within each product code will then determine the submission pathway — either a premarket notification, premarket approval, PMA, or de novo if a product code does not exist for a software device. The means of compliance is typically based on clinical experience, evidence, trials and on demonstrating substantial equivalence to a predicate device.
While these two frameworks form the basis of the SaMD regulatory pathway, each jurisdiction has its own set of requirements.
The US Regulatory Framework
The US FDA has been at the forefront in trying to address the challenges of regulating SaMDs, with the introduction of the 21st Century Cures Act back in 2016. In particular, the Act included a section that adds a definition of a SaMDs in line with IMDRF recommendations.
As a result, a number of changes were made to the regulations, including to off-the-shelf software use, cybersecurity, as well as new guidance documents specifically for medical device software, such as guidance for mobile medical applications, general wellness devices, medical device data systems, artificial intelligence and machine learning.
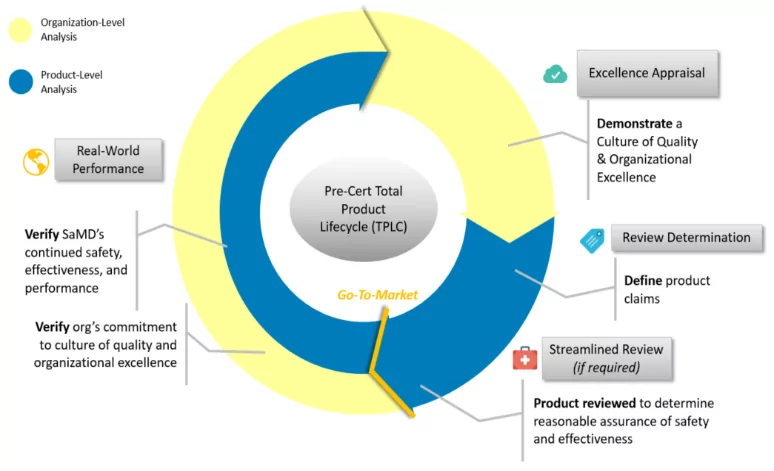
One of the new initiatives that came out of the 21st Century Cures Act was a launch of the software pre-certification pilot program aimed at helping to inform the development of a future regulatory model that will provide more streamlined and efficient regulatory oversight of software-based medical devices developed by manufacturers who have demonstrated a robust culture of quality and organizational excellence, and who are committed to monitoring real-world performance of their products. The focus of the program is on a total product lifecycle approach, as opposed to pre-market applications.
FDA’s Pilot Digital Health PreCert Total Lifecycle Approach
The EU Regulatory Framework
In May 2021, the new Medical Devices Regulation came into effect, which includes significant changes for SaMDs, with clear definitions of what an SaMD is, as opposed to software intended for lifestyle and wellbeing. The In Vitro Diagnostic Regulation (IVDR) also includes stand-alone software as an IVD device, which is treated like any other IVD device, including classification and regulatory options and requirements.
With the MDR in place, most software previously classed as class one under the Medical Devices Directive is likely to be up-classified to class IIa or higher. Other changes include recognition that software can be regarded as an active device, inclusion of requirements around software development lifecycle, information security and mobile computing platforms, and special labelling requirements.
In addition, in 2019 the Medical Device Coordination Group in Europe published guidance on qualification and classification of software for the MDR and IVDR. The guidance provides definitions and criteria around SaMDs, as well as greater clarity and examples of how to apply the new rules for various types of software.
Australia’s Regulatory Framework
As of February 2021, the Therapeutic Goods Administration (TGA) has implemented reforms to the regulation of SaMDs. One of the significant changes was the introduction of new classification rules. Most devices will remain in the same class if they provide direct diagnosis or monitoring. Devices intended to provide therapy by the provision of information, for example, a medical device intended to provide cognitive behavioral therapy, could potentially be classified as a higher class.
There have also been a number of changes to the essential principles, such as with regards to the management of data and information as it applies to cyber security, and requirements relating to development, production and maintenance. One amendment allows information, where applicable, to be provided electronically, rather than on a leaflet, for SaMDs. The TGA has also released software guidance documents for excluded or exempt devices for certain types of SaMDs.
Canada’s Regulatory Framework
In December 2019, Health Canada published a guidance document on SaMDs and adopted the IMDRF framework. Under the new guidance, low risk software is classified as class I and higher risk software is classified as class III, based on the significance of the information provided by the SaMD to the healthcare decision, and the state of patient’s healthcare situation or condition.
The Health Canada guidance also covers exclusion criteria for certain types of SaMDs.
Staying ahead of regulatory change
The rapidly evolving nature of software technologies inevitably has a knock-on effect on regulations as health authorities scramble to ensure the safety of patients. But, while staying ahead of change can be difficult for SaMD manufacturers, the majority of new regulations go through a public consultation period, which gives the industry an opportunity to provide feedback and influence the regulation. It’s important, therefore, that companies stay abreast of new regulatory developments, take part in the consultations, and provide their feedback.



